
Las células de la cresta neural (CCN) son una población de células madre transitorias, caracterizadas por su gran capacidad migratoria y pluripotencialidad.
La cresta neural es exclusiva de los vertebrados y se divide en cuatro subpoblaciones: craneal, cardíaca, vagal y troncal. Cada subpoblación tiene la capacidad de diferenciarse en células diversas y especializadas, y en tejidos particulares del nivel axial de origen.1
Las células de la cresta neural craneal (CCNC) se encuentran adyacentes a la región del cerebro medio/trasero en desarrollo y en última instancia, migran a los arcos branquiales, donde se diferencian en la mayoría de los huesos, tejidos conectivos y ciertos nervios de la cabeza.2
El desarrollo craneofacial normal es consecuencia de complejas interacciones en el tejido embrionario, las cuales requieren de una regulación precisa del movimiento celular, crecimiento, patrón y diferenciación de los tejidos craneofaciales. Múltiples estudios genéticos han determinado la participación de numerosos genes en este proceso, incluyendo genes que codifican factores de transcripción, factores de crecimiento y receptores.3
Uno de los defectos de nacimiento más frecuentes son las anomalías craneofaciales, causadas por la combinación entre factores genéticos, como mutaciones en los genes involucrados, y factores ambientales, que pueden incluir la exposición de la madre a toxinas del tabaco, alcohol o ciertos medicamentos.1 Como resultado, se generan anormalidades como hendiduras faciales y craneosinostosis.3
Gracias a los avances tecnológicos y científicos, se han podido identificar las interacciones moleculares y los puntos de regulación en el desarrollo de las CCNC. En conjunto, estos factores reguladores y sus conexiones se agrupan en una gran Red Reguladora de Genes (RRG) que dirige el proceso.2
Dentro de los factores involucrados en el correcto desarrollo craneofacial, destacan los que pertenecen a la familia de genes homeóticos o Hox, siendo los genes Msx (Msx1, Msx2 y Msx3) importantes representantes de la familia, identificados como homólogos de los genes homeóticos de Drosophila melanogaster. Estos genes codifican las proteínas Msx, las cuales son importantes reguladores del desarrollo craneofacial, del sistema linfático y sistema nervioso.3
Los genes Hox codifican una amplia familia de factores de transcripción caracterizados por poseer un homeodominio en su estructura. Este homeodominio se conforma por una secuencia de unión al DNA, muy conservada a través de la evolución, la cual está constituida por 61 aminoácidos formando tres a-hélices. Los genes Hox se caracterizan por su activación en cascada, es decir, las proteínas codificadas por los primeros genes regulan la expresión de los genes posteriores.4
Durante el desarrollo del cráneo, Msx1 y Msx2 se expresan en el mesénquima de la sutura y la dura madre. Mientras que la expresión de Msx1 se extiende a estadios postnatales de morfogénesis, Msx2 evidencia una abrupta disminución en su expresión posterior al nacimiento. Adicionalmente, Msx1 se expresa durante el desarrollo dentario, hasta el momento previo a la diferenciación de odontoblastos y ameloblastos.3
Como parte de la red reguladora de genes, los genes Hox interactúan con una gran cantidad de genes adicionales, entrelazando sus vías de señalización. Por ejemplo, se ha reportado que la vía de señalización de los Hedgehog (Hh), activada por las proteínas Sonic Hedgehog (SHH), muestra un importante papel en el desarrollo de varios órganos, incluyendo tejido craneofacial como paladar, labio, glándulas salivales y dientes.5
Por su parte, también participan los genes BMP, los cuales codifican para proteínas morfogenéticas óseas (BMP), las cuales son miembros de la súperfamilia del Factor de Crecimiento Transformante b (TGF-b). Las BMP participan en el desarrollo de múltiples tejidos, incluyendo el desarrollo craneofacial y se expresan en regiones discretas de las zonas distales del inicio del primordio facial y en la formación del cráneo y las meninges, siendo regulados por los antagonistas Chordin o Noggin. De las BMP identificadas, BMP2, 4, 6, 7, 9 tienen funciones osteogénicas, BMP3 es un regulador negativo de la densidad ósea y BMP13 es un fuerte inhibidor de la formación ósea.6,7
Otro grupo importante de genes, son los Wnt, los cuales intervienen en las funciones de las comunicaciones intercelulares durante el desarrollo de los órganos y de los miembros del embrión. Participan en procesos de proliferación celular, diferenciación, migración y patrones celulares, además de la morfogénesis craneofacial, interactuando con los genes MSX1 y BMP4.7
Alteraciones en cualquiera de estos genes, por mutaciones, incersiones, deleciones, o simplemente, la amplificación de los mismos, se asocia con múltiples manifestaciones, las cuales pueden ser evidenciadas en la práctica clínica odontológica. Alteraciones en la función de Msx1 se asocia con hipoplasias mandibulares y maxilares, fisuras labiales con o sin fisura palatina, alteraciones frontonasales, hipodoncias y alteraciones en el desarrollo de las extremidades y del sistema nervioso. El Síndrome de Witkop y el Síndrome de Wolf-Hirschhorn (Fotografía 1) se vinculan con esta alteración.3

Fotografía 1.- Vista frontal de niños con Síndrome de Wolf-Hirschhorn, mostrando grados variables del compromiso craneofacial.8
En el caso de Msx2, la alteración de su función se asocia directamente con la Craneostosis tipo Boston (Fotografía 2), debido a una sustitución de una prolina, por una histidina, en la posición 7 del exón 1, del homeodominio. Sus manifestaciones son obliteración de la sutura del cráneo, hipoplasia media facial, anormalidades en el crecimiento de la base craneal y alteraciones en la cara y en las extremidades.3
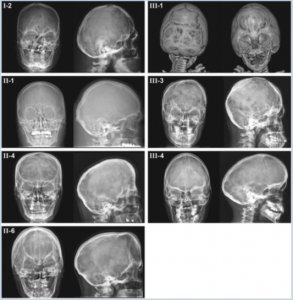
Fotografía 2.- Imágenes radiográficas (anterior, lateral y posterior) del cráneo de individuos con craneosinostosis tipo Boston.9
La pérdida de SHH, produce defectos en el patrón de la placa neural: prosencefalia, defectos análogos al hipertelorismo y labio-paladar hendido, puente nasal plano y microcefalia. La Holoprosencefalia no cromosómica, no sindrómica (Fotografía 3), muestra un patrón de herencia autosómica dominante, con una penetrancia incompleta.10
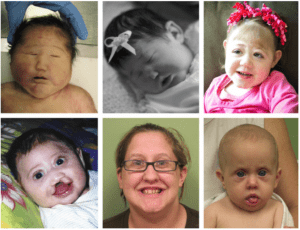
Fotografía 3.- Individuos con mutaciones en el gen SHH, asociadas con Holoprosencefalia.10
Existen numerosos genes relacionados con la morfogénesis craneofacial, y cada día la investigación en esta área crece y con ello, el número de genes reportados en la literatura. Las complejas vías de señalización dificultan la manipulación de los procesos patogénicos, asociados con malformaciones craneofaciales. Adicionalmente, existe un solapamiento de los fenotipos en los distintos síndromes, dificultando la identificación de factores de riesgo ambientales. La identificación de nuevas mutaciones y la continua evidencia científica permitirá mejorar el cribado prenatal y la consejería genética de estos pacientes.

